REV ARGENT NEUROC. VOL. 34, N° 3: 207-213 | 2020
ARTÍCULOS DE REVISIÓN
Principales biomarcadores moleculares en la caracterización de los tumores del sistema nervioso central
Alejandra Báez,1 Alejandra Gonzalez Roffo,1 José Gómez Escalante,2 Álvaro Campero,3
Ignacio Casas Parera1
1Departamento de Neurología, Centro Universitario de Neurooncología. Instituto de Oncología Ángel H. Roffo. C.A.B.A.
2Departamento de Patología, Instituto de Oncología Ángel H. Roffo. C.A.B.A.
3Servicio de Neurocirugía, Hospital Ángel C. Padilla. Tucumán.
RESUMEN
La Clasificación de Tumores del Sistema Nervioso Central de la OMS 2016 incorpora biomarcadores moleculares junto a las características histológicas clásicas, en un diagnóstico integrado, con el fin de definir distintas entidades de gliomas con la mayor precisión posible. Los estudios de perfiles moleculares en el genoma han revelado las alteraciones genéticas características y los perfiles epigenéticos asociados con diferentes tipos de gliomas. Estas características moleculares pueden usarse para refinar la clasificación del glioma, mejorar la predicción de los resultados obtenidos con los tratamientos actuales y futuros en los pacientes, y como guía de un tratamiento personalizado. Asimismo, tener una aproximación pronóstica en cada paciente. Este cambio de paradigma ha modificado la forma en que se diagnostica el glioma y sus implicancias en la práctica diaria en la indicación de los diferentes tratamientos al paciente. Aquí, sintéticamente, revisamos y destacamos los biomarcadores moleculares clínicamente relevantes. Intentamos dejar plasmado cómo los avances en la genética molecular de los gliomas pueden promover y allanar el camino hacia la medicina de precisión en neurooncología.
Palabras claves: Glioma; Biomarcadores Moleculares; Mutaciones; Clasificación OMS 2016; Neurooncología
ABSTRACT
The Classification of Tumors of the Central Nervous System of the WHO 2016 incorporates molecular biomarkers together with the classical histological characteristics, in an integrated diagnosis, in order to define different glioma entities with the highest possible accuracy. Studies of molecular profiles in the genome have revealed characteristic genetic alterations and epigenetic profiles associated with different types of gliomas. These molecular characteristics can be used to refine the classification of gliomas, improve the prediction of the results obtained with current and future treatments in patients and as a guide for a personalized treatment. Also, have a prognostic approach in each patient. This paradigm shift has modified the way glioma is diagnosed and its implications in daily practice in the indication of different treatments to the patient. Here, synthetically, we review and highlight clinically relevant molecular biomarkers. We try to capture how advances in the molecular genetics of gliomas can promote and pave the way to precision medicine in neuro-oncology.
Key words: Glioma; Molecular Biomarkers; Mutations; WHO Classification 2016; Neuro-Oncology
INTRODUCCIÓN
Los tumores primarios del sistema nervioso central (SNC) abarcan diversos tipos que incluyen los derivados del parénquima, vainas de nervios craneales o de las cubiertas meníngeas; con una prevalencia variable según la edad del paciente y la ubicación del tumor. Los tumores primarios malignos en orden de frecuencia son los glioblastomas, astrocitomas grado III (GIII), los oligodendrogliomas anaplásicos -GIII-, ependimomas RELA fusión-positivo y anaplásicos, entre otros. En conjunto, los gliomas representan aproximadamente el 75% de los tumores primarios malignos del SNC.17,28
Hasta hace unos años la clasificación de los tumores del SNC se basaba exclusivamente en las características morfológicas y el grado de diferenciación celular.
Los problemas diagnósticos en neuropatología son ampliamente reconocidos: las discordancias diagnósticas entre diferentes observadores, el análisis histológico y la clasificación debido a la naturaleza inherentemente subjetiva de ciertos aspectos de la interpretación histopatológica. Además, la muestra de biopsia no siempre es representativa y puede no presentar todas las características diagnósticas relevantes en tumores con morfología y biología heterogéneas.
A partir de los ’90 hubo una comprensión más acabada sobre la patogénesis y los perfiles moleculares de estos tumores, que permitió separarlos -aun cuando a la microscopía eran similares- con una mayor precisión en distintos subgrupos y con comportamiento clínico similar.
Estos cambios impulsaron la publicación en el año 2016 de una actualización en la clasificación de los tumores del SNC de la OMS, donde los criterios histológicos tradicionales se complementaron con biomarcadores genómicos (WHO CNS 2016).
En este trabajo solo se incluirán los marcadores moleculares aplicados al diagnóstico de los principales tumores de estirpe glial.13
Esta caracterización genética/molecular complementa el análisis histológico estándar, proporcionando información diagnóstica, predictiva y pronóstica adicional, con una significativa mejora en la tipificación tumoral final, e influir en la selección del tratamiento y una mejora en la toma de decisiones en estos pacientes.
OBJETIVOS
Describir los principales biomarcadores que se recomiendan actualmente en neurooncología para el estudio de los gliomas.
MATERIALES Y MÉTODOS
Se realizó revisión de la bibliografía a través de PubMed utilizando como criterios de búsqueda artículos en revistas utilizando las palabras claves: glioma; biomarcadores moleculares; mutaciones; clasificación OMS 2016; neurooncología, en el período 2000-2019, y el libro de la OMS (2016) sobre la clasificación de los tumores del sistema nervioso central. Se realizó revisión de la historia clínica del caso presentado.
RESULTADOS
A continuación, se describen los marcadores más utilizados en la práctica para el diagnóstico biomolecular de los gliomas. Se seleccionaron 54 artículos, de los cuales por antecedentes históricos y graduales avances, se incorporaron al presente trabajo 28 además del libro de las OMS 2016.
Mutación de IDH
La IDH (isocitrato deshidrogenasa) es una enzima involucrada en el proceso del metabolismo celular y participa en la respuesta al estrés oxidativo. Existen 3 isoformas (IDH 1, 2 y 3). En la práctica se analizan las mutaciones de IDH1 y eventualmente IDH2. Estas mutaciones determinan la pérdida de su función.9
Las mutaciones de IDH1 (395G→A: residuo Arg 132) e IDH2(515G→A: residuos Arg 172) inhiben la función de la enzima IDH, dando lugar a la generación de 2 hidroxiglutarato (2-HG), un oncometabolito que origina una disminución en los niveles de ɑ-cetoglutarato y un incremento en los niveles de radicales libres. Como consecuencia, se alteran tanto la actividad metabólica celular como la mitosis, y al aumentar la metilación del ADN favorecen la aparición de nuevas mutaciones.1,19
La mutación IDH1 más común es en el codón R132H, y se analiza mediante técnicas inmunohistoquímicas. Si la mutación R132H es negativa, pero existe una alta sospecha clínica, se recomienda la secuenciación de IDH1 e IDH2 para detectar mutaciones menos frecuentes.9
Estas mutaciones se han descripto principalmente entre el 60% y el 80% de los gliomas de bajo grado y en el 75% de los glioblastomas secundarios. Esta mutación no está presente en patologías que pueden imitar a un glioma, tales como vasculitis, encefalitis, enfermedad desmielinizante o la gliosis reactiva.9,16
Las mutaciones en los genes IDH1 e IDH2 se asocian con una mayor respuesta a la quimioterapia, mejor pronóstico y mayor supervivencia global libre de progresión.1,4
El IDH es requisito para el diagnóstico de oligodendroglioma. Esta mutación puede estar presente o no en los astrocitomas. Cuando se encuentra mutada en el glioblastoma es diagnóstico de glioblastoma secundario, este último con mejor pronóstico que el glioblastoma de novo.16
Codeleción 1p/19q
Esta codeleción corresponde a una traslocación balanceada, donde se pierde el brazo corto del cromosoma 1 y el brazo largo del cromosoma 19.
Independientemente de los hallazgos histológicos, la presencia de codeleción1p/19q asociada a mutación de IDH hace diagnóstico de oligodendroglioma.26,27
La presencia de la codeleción 1p/19q está fuertemente asociada con la histología oligodendroglial y ayuda a confirmar el diagnóstico en tumores con características histológicas ambiguas o mixtas. Los gliomas con IDH mutado que no muestren pérdida de expresión de ATRX (gen del síndrome de déficit intelectual/alfa talasemia relacionado al X), deben ser testeados para la búsqueda de codeleción 1p/19q, incluso aquellos que no muestren una histología clara de oligodendroglioma.2,12
La detección de codeleción 1p/19q no es necesaria si un glioma es IDH no mutado. Es decir, el glioma solo puede considerarse oligodendroglioma si presenta codeleción 1p/19q asociado a la mutación de IDH.22
El análisis de la codeleción 1p/19q se realiza por técnica de hibridación in situ (FISH) o reacción en cadena de la polimerasa (PCR). También se pueden emplear métodos adicionales. La presencia de codeleción 1p/19q es predictiva de mejor respuesta a la quimioterapia con agentes alquilantes y la terapia combinada (radioterapia asociada a quimioterapia), y por ende de un mejor pronóstico.3,25
Metilación del promotor de MGMT
La enzima (O6-metilguanina-ADN metiltransferasa) es una enzima reparadora del ADN que revierte el daño causado por los agentes alquilantes, y por ello genera resistencia tumoral al tratamiento. La metilación del promotor de la enzima silencia su actividad (la inactiva), haciendo que el tumor sea más sensible a los agentes alquilantes.6
El análisis de la metilación del promotor de se realiza por técnicas de PCR (metilación específica por pirosecuencia), o técnicas basadas en matrices. La metilación del promotor de la enzima debería formar parte esencial del diagnóstico molecular en los gliomas de alto grado por su valor predictivo y pronóstico.29
Esta alteración epigenética se encuentra en el 35-50% de los glioblastomas. El glioblastoma con promotor de la metilado se asocia con una mejor respuesta al tratamiento con radioterapia concomitante con temozolomida, y mayor supervivencia.5,7
En pacientes añosos con gliomas de alto grado, la presencia de metilado resultaría de utilidad para definir la conducta terapéutica, ya que los que presenten esta mutación se beneficiarán del tratamiento con agentes alquilantes.7,14
Mutación de ATRX
El gen ATRX (gen del síndrome de déficit intelectual/alfa talasemia relacionado al X) codifica una proteína reguladora de la cromatina. La inactivación del gen puede ser por mutación, deleción o fusión génica. Esta inactivación del gen ATRX se asocia a la presencia de las mutaciones de IDH y del gen TP53, presente en los astrocitomas. La presencia de mutación -déficit- de ATRX es casi mutuamente excluyente con la codeleción 1p/19q.10,15
La deficiencia de ATRX junto con la mutación de IDH es típica del astrocitoma.21
Mutación del TP53
El gen TP53 es un gen supresor tumoral. La mutación del TP53 está presente en el 30-50% de los astrocitomas, y suele estar asociado al déficit de ATRX. Los glioblastomas que presentan mutación del TP53 son de observación en pacientes jóvenes con diagnóstico previo de gliomas de bajo grado (glioblastoma 2rio.).5
Amplificación del EGFR
El receptor de factor de crecimiento epidérmico (EGFR) se localiza en el cromosoma 7. La ganancia del cromosoma 7p combinada con la pérdida del cromosoma 10q es la alteración genética más frecuente en el glioblastoma (presente en aproximadamente el 50%), y la mayor parte de las veces la amplificación de EGFR ocurre en los gliomas que presentan estas alteraciones cromosómicas. La amplificación del EGFR está presente en el 40% de los glioblastomas primarios. Esta alteración facilita la replicación celular incontrolada.5,11
Mutación de la Histona H3K27M
En un alto porcentaje de los gliomas difusos de línea media (antes "tumores de tronco" o "gliomas pontinos") se encuentra presente la mutación H3K27M. Esta mutación ha sido identificada en pacientes de grupo etario variable, y también en tumores del tercer ventrículo, región pineal, cerebelo y médula espinal. La determinación de esta mutación debe solicitarse en un contexto clínico-radiológico adecuado.23
Cuando se publicó la clasificación neuropatológica OMS 2016, se pensaba que la mutación H3K27M era específica para gliomas difusos de la línea media. Sin embargo, actualmente se han reportado mutaciones H3K27M en gliomas circunscriptos de la línea media como ganglioglioma, astrocitoma pilocítico, tumores glioneuronales no específicos y ependimomas. Además, la mutación H3K27M puede coexistir con mutación de BRAF V600E (es una serina/treonina kinasa que activa la vía de transducción de las MAP cinasas que participan en la diferenciación y proliferación celular). Esto significa que la presencia de una mutación H3K27M no necesariamente define el diagnóstico de glioma difuso de la línea media. El contexto histológico tiene importancia; los tumores deben ser definitivamente de línea media, ser gliomas difusos infiltrantes y con la mutación presente, para un diagnóstico de certeza.18,28
La presencia de esta mutación en los gliomas difusos de la línea media es de mal pronóstico y se corresponde con el grado IV de la OMS.24
Mutación de BRAF
La fusión y/o mutación BRAF (variante V600E) está comúnmente presente en tumores como melanoma, pero además se encuentra entre el 60% y el 80% de los xantoastrocitomas pleomórficos supratentoriales (grado II-III), 30% de los tumores glioneuronales, 20% de los gangliogliomas y en un 5% de los astrocitomas pilocíticos.8,20
Los tumores BRAF V600E pueden responder a los inhibidores de BRAF como vemurafenib; se continúan los ensayos clínicos. El análisis de la mutación de BRAF debe realizarse en un contexto clínico-radiológico apropiado. En la Tabla se muestra la correlación entre algunos tumores primarios del SNC y marcadores moleculares.
Seguidamente, un caso clínico que muestra la aplicación práctica de estos biomarcadores.
Tabla: Correlación entre algunos tumores y biomarcadores moleculares13
|
Mutación de IDH |
Codeleción 1p/19q |
Mutación de ATRX |
Mutación de TP53 |
Metilación de MGMT |
Amplificación de EGFR |
Mutación H3K27M |
Mutación de BRAF |
| Astrocitoma IDH mutado |
+ |
- |
0,86 |
0,94 |
Infrecuente |
Infrecuente |
No aplica |
No aplica |
| Astrocitoma IDH no mutado |
-(*) |
- |
Infrecuente |
Infrecuente |
+ (50% símil GB) |
+ (35%) símil GB |
Infrecuente |
Infrecuente |
| Oligodendroglioma |
+ |
+ |
- |
- |
Infrecuente |
- |
No aplica |
No aplica |
| Glioblastoma (GB) |
- |
- |
Infrecuente |
0,27 |
0,5 |
+ (hasta en un 50%) |
No aplica |
No aplica |
| Glioblastoma secundario |
+ |
- |
0,71 |
0,81 |
+ |
Infrecuente |
No aplica |
No aplica |
| Gliomas de línea media |
- |
Infrecuente |
14,75 |
29,5 |
Desconocido |
- |
0,8 |
No aplica |
| Xantoastrocitoma |
- |
- |
- |
Infrecuente |
- |
- |
No aplica |
+(**) |
* Agotar las restantes mutaciones de IDH1, y finalmente IDH2.
** Esta mutación también puede observarse en otros tumores como: astrocitoma pilocítico, ganglioglioma, astrocitoma subependimario de células gigantes y tumores glioneuronales.
CASO CLÍNICO
Paciente de 49 años de edad, que en noviembre de 2012 presentó crisis de inicio focal sin alteración de la consciencia de tipo autonómica. Las neuroimágenes mostraron una lesión temporoparietal derecha (fig. 1 A-D). Inició tratamiento con fármacos antiepilépticos -DFH- y por rash pasó a levetiracetam. En julio de 2013 se realizó resección parcial de la lesión (área elocuente). La anatomía patológica (AP) informó: oligodendroglioma GII, 1p19q con deleción parcial. Continuó sin tratamiento oncoespecífico y con controles por neuroimágenes. En 2018 concurrió al Instituto Roffo para continuar tratamiento. El examen neurológico fue irrelevante; continuaba con crisis focales. Se solicitó nueva neuroimagen y se ajustó dosis de levetiracetam. La revisión AP (fig. 2 A-F) informó hallazgos morfológicos clásicos de oligodendroglioma, pero con un perfil inmunohistoquímico: ausencia de codeleción 1p19q, IDH1 mutado, ATRX negativa (mutada), sobreexpresión del TP53 y un Ki67 2% (bajo), en relación a un fenotipo astrocítico; corresponde a un astrocitoma difuso IDH1 mutado. Se indicó tratamiento quimioterápico con temozolomida en ciclos de 5 días consecutivos c/28 días. La determinación de no estaba disponible al momento del caso clínico.
El análisis biomolecular de la revisión anatomopatológica permitió obtener un diagnóstico preciso que posibilitó adecuar la conducta terapéutica.
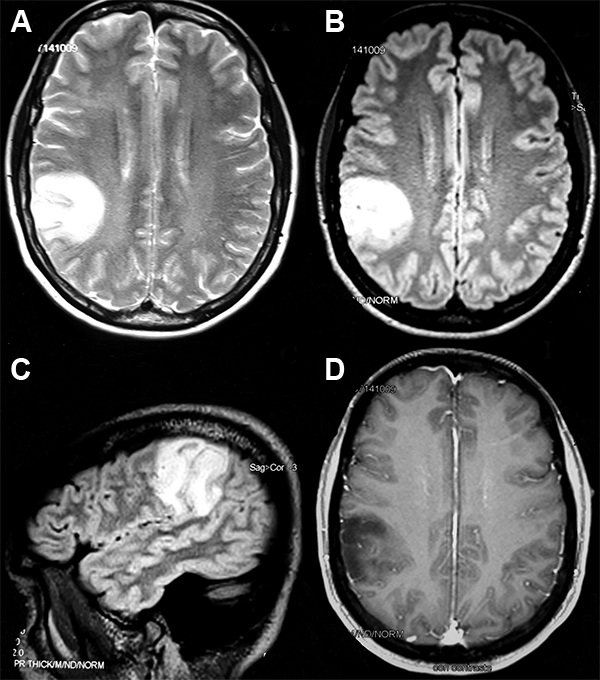
Figura 1: RM de cerebro (cortes axiales en secuencia T2 -A- y FLAIR -B y C-). Lesión de bordes nítidos, corticosubcortical frontoparietal perirrolándica derecha, hiperintensa, con efecto de masa local y colapso de surcos. D) Corte axial (secuencia T1c/ gadolinio). La lesión se muestra heterógénea iso/hipointensa con muy tenue realce tras la administración del contraste.
Figura 2: A) Tinción H&E: proliferación neoplásica de baja densidad celular constituida por núcleos esféricos a ovales y escaso citoplasma acidófilo, con halo claro perinuclear. B) GFAP: positivo en astrocitos reactivos y oligodendrocitos gliofibrilares. C) IDH1: positivo en células neoplásicas (mutado). D) ATRX: negativo en elevado porcentaje (proteína mutada). E) TP53: marcación en elevado porcentaje (80%). F) Ki 67: índice del 2%.
DISCUSIÓN
El diagnóstico y tipificación de los tumores primarios del SNC debe realizarse basados en estos marcadores biomoleculares, y asociado a la evaluación histológica estándar. Todos ellos se realizan en el Instituto Roffo de la Universidad de Buenos Aires. Aquéllos nos permiten distinguir y diferenciar con mayor precisión a estos tumores, con un conocimiento más certero de la probable respuesta a los tratamientos actuales, y del comportamiento y evolución de éstos (factores predictivo y pronóstico). En el caso presentado, al ser un glioma de bajo grado, el valor de la determinación de la metilada es relativo. Su uso y utilidad, sobre todo, se reserva para los gliomas de alto grado.
CONCLUSIONES
Estos diagnósticos moleculares, en la actualidad y en un futuro, abren un abanico de tratamientos que permiten un mejor control tumoral, y mayor calidad y sobrevida de los pacientes.
BIBLIOGRAFÍA
- Borodovsky A, Seltzer MJ, Riggins GJ. Altered cancer cell metabolism in gliomas with mutant IDH1 or IDH2. Curr Opin Oncol. 2012;24(1):83-9.
- Burger PC, Minn AY, Smith JS, Borell TJ, Jedlicka AE, Huntley BK et al. Losses of chromosomal arms 1p and 19q in the diagnosis of oligodendroglioma. A study of paraffin-embedded sections. Mod Pathol. 2001;14(9):842-53.
- Cairncross G, Wang M, Shaw E, Jenkins R, Brachman D, Buckner J et al. Phase III trial of chemoradiotherapy for anaplastic oligodendroglioma: long-term results of RTOG 9402. J Clin Oncol. 2013;31(3):337-43.
- de Quintana-Schmidt C, Alvarez-Holzapfel MJ, Nomdedeu-Guinot J, Bague-Rosell S, Gallego-Rubio O, Leidinger A et al. Isocitrate dehydrogenase type I mutation as a prognostic factor in glioblastoma and a literature review. Neurocirugía (Astur). 2015;26(6):276-83.
- Diamandis P, Aldape KD. Insights from molecular profiling of adult glioma. J Clin Oncol. 2017;35(21):2386-93.
- Esteller M, Garcia-Foncillas J, Andion E, Goodman SN, Hidalgo OF, Vanaclocha V et al. Inactivation of the DNA-repair gene and the clinical response of gliomas to alkylating agents. N Engl J Med. 2000;343(19):1350-4.
- Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M et al. gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997-1003.
- Horbinski C. What do we know about IDH1/2 mutations so far, and how do we use it? Acta Neuropathol. 2013;125(5):621-36.
- Horbinski C. To BRAF or not to BRAF: is that even a question anymore?. J Neuropathol Exp Neurol. 2013;72(1):2-7.
- Koschmann C, Calinescu AA, Nunez FJ, Mackay A, Fazal-Salom J, Thomas D, et al. ATRX loss promotes tumor growth and impairs non-homologous end joining DNA repair in glioma. SciTransl Med. 2016;8(328):328ra28.
- Koshiyama DB, Trevisan P, Graziadio C, Rosa RFM, Cunegatto B, Scholl J et al. Frequency and clinical significance of chromosome 7 and 10 aneuploidies, amplification of the EGFR gene, deletion of PTEN and TP53 genes, and 1p/19q deficiency in a sample of adult patients diagnosed with glioblastoma from Southern Brazil. J Neurooncol.2017;135(3):465-72.
- Labussiere M, Idbaih A, Wang XW, Marie Y, Boisselier B, Falet C et al. All the 1p19q codeleted gliomas are mutated on IDH1 or IDH2. Neurology. 2010;74(23):1886-90.
- Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Ellison DW, Figarella-Brangec D, Perry A, Reifenberger G, von Deimling A, Eds. WHO Classification of Tumours of the Central Nervous System. Fourth Edition. Lyon, France: IARC Press; 2016.
- Malmstrom A, Gronberg BH, Marosi C, Stupp R, Frappaz D, Schultz H et al. Temozolomide versus standard 6-week radiotherapy versus hypofractionated radiotherapy in patients older than 60 years with glioblastoma: the Nordic randomised, phase 3 trial. Lancet Oncol. 2012;13(9):916-26.
- Nandakumar P, Mansouri A, Das S. The role of ATRX in glioma biology. Front Oncol. 2017;7:236.
- Ohgaki H, Kleihues P. The definition of primary and secondary glioblastoma. Clin Cancer Res. 2013;19(4):764-72.
- Ostrom QT, Gittleman H, Truitt G, Boscia A, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2011–2015. Neuro-Oncology. 2018;20(4):1-86.
- Pages M, Beccaria K, Boddaert N, Saffroy R, Besnard A, Castel D et al. Co- occurrence of histone H3 K27M and BRAF V600E mutations in paediatric midline grade I ganglioglioma. Brain Pathol. 2018;28(1):103-11.
- Prensner JR, Chinnaiyan AM. Metabolism unhinged: IDH mutations in cancer. Nat Med. 2011;17(3):291-3.
- Qaddoumi I, Orisme W, Wen J, Santiago T, Gupta K, Dalton JD et al. Genetic alterations in uncommon low-grade neuroepithelial tumors: BRAF, FGFR1, and MYB mutations occur at high frequency and align with morphology. ActaNeuropathol. 2016;131(6):833-45.
- Reuss DE, Sahm F, Schrimpf D, Wiestler B, Capper D, Koelsche C et al. ATRX and IDH1-R132H immunohistochemistry with subsequent copy number analysis and IDH sequencing as a basis for an "integrated" diagnostic approach for adult astrocytoma, oligodendroglioma and glioblastoma. Acta Neuropathol. 2015;129(1):133-46.
- Sahm F, Reuss D, Koelsche C, Capper D, Schittenhelm J, Heim S et al. Farewell to oligoastrocytoma: in situ molecular genetics favor classification as either oligodendroglioma or astrocytoma. Acta Neuropathol. 2014;128(4):551-9.
- Solomon DA, Wood MD, Tihan T, Bollen AW, Gupta N, Phillips JJ et al. Diffuse midline gliomas with histone H3-K27M mutation: a series of 47 cases assessing the Spectrum of morphologic variation and associated genetic alterations. Brain Pathol. 2016;26(5):569-80.
- Sturm D, Witt H, Hovestadt V, Khuong-Quang DA, Jones DT, Konermann C et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 2012;22(4):425-37.
- van den Bent MJ, Brandes AA, Taphoorn MJ, Kros JM, Kouwenhoven MC, Delattre JY et al. Adjuvant procarbazine, lomustine, and vincristine chemotherapy in newly diagnosed anaplastic oligodendroglioma: long-term follow-up of EORTC brain tumor group study 26951. J Clin Oncol. 2013;31(3):344-50.
- van den Bent MJ, Smits M, Kros JM, Chang SM. Difusse infiltrating oligodendroglioma and astrocytoma. J Clin Oncol 2017; 35(21):2394-401.
- Weller M, Stupp R, Hegi MG, van den Bent M, Tonn JC, Sanson M et al. Personalized care in neuro-oncology coming of age: why we need and 1p/19q testing for malignant glioma patients in clinical practice. Neuro Oncol. 2012;14(4):100-8.
- Wood MD, Halfpenny AM, Moore SR. Applications of molecular neuro-oncology - a review of diffuse glioma integrated diagnosis and emerging molecular entities. Diagn Pathol. 2019;14(1):29.
- Xie H, Tubbs R, Yang B. Detection of promoter methylation in glioblastoma using pyrosequencing. Int J Clin Exp Pathol. 2015;8(1):636-42.
COMENTARIO
Los autores presentan una comunicación con formato de revisión no sistemática acerca de los principales biomarcadores moleculares en la caracterización de los tumores del sistema nervioso central, en particular de las neoplasias de origen glial (gliomas).
En los últimos 20 años la investigación básica en el campo de la tumorogénesis y el desarrollo tumoral, ha reportado alteraciones genéticas y epigenéticas que determinan subgrupos de proliferaciones con diferente evolución (marcadores moleculares pronósticos) y subgrupos con diferente respuesta a tratamientos (marcadores moleculares predictivos). Por este hecho, la clasificación de tumores del sistema nervioso central de la OMS versión 2016 introduce por primera vez la necesidad de contar con la realización de estudios de biología molecular en algunos tumores del SNC, para su categorización.
Asimismo, la introducción del uso de estos marcadores permite contar con elementos objetivos de diagnóstico, reduciendo la variabilidad interobservador habitual, incluso entre patólogos entrenados.
Si bien el tratamiento principal de los gliomas ha sido - y continúa siendo - el mayor grado de resección quirúrgica posible con preservación de la función, este nuevo enfoque paradigmático intenta definir e individualizar la “personalidad” de estas lesiones, permitiendo diseñar mejores protocolos terapéuticos, a la vez que conocer con mayor precisión su evolución pronóstica.
Agradecemos a los autores.
Claudio Centurión
Sanatorio Aconcagua. Córdoba
COMENTARIO
El conocimiento de los marcadores moleculares precisan diagnóstico, respuesta a la quimioterapia y pronóstico, y surgen como resultado de una rápida incorporación de conocimientos que buscan comprender la génesis tumoral. Considerar que la mutación en el gen de la IDH en gliomas fue identificada en el 2009 y hoy es la protagonista de la clasificación de la OMS 2016, refleja un vertiginoso cambio en la manera de concebir la enfermedad. Marcadores que también pueden servir como blanco de una futura terapia génica. Aunque la utilidad clínica está en estudio y el impacto en la sobrevida aún es austero, los estudios moleculares son un paso importante para entender los mecanismos biológicos que gobiernan el desarrollo de los gliomas.
Tomás Funes
Sanatorio Anchorena. C.A.B.A. Argentina
COMENTARIO
Los autores realizan una correcta reseña sobre la actualización en la clasificación de gliomas difusos publicada por la OMS en 2016. Agregaría que la misma, desprende escritos para identificar aquellos tumores que no entran en la clasificación habiendo sido estudiados con técnicas moleculares.1 También, destacar que la población pediátrica plantea otro escenario2 y que la topografía del tumor es un dato fundamental.
Enumeran claramente los marcadores moleculares disponibles por inmunohistoquímica y biología molecular señalando su utilidad en cada grupo.
El caso presentado ejemplifica el algoritmo del diagnóstico integrado en gliomas difusos de bajo grado, y sus implicancias en el pronóstico y tratamiento a largo plazo. Las fotos histológicas son ilustrativas y acertadas. A modo personal comento que con la hematoxilina eosina (fig. 2A) podemos apreciar la superposición de criterios morfológicos que limitan el diagnóstico histológico.
Felicito a los autores por el aporte y el trabajo realizado.
Silvina Figurelli
Hospital Dr. Juan A. Fernández. C.A.B.A., Buenos Aires
BIBLIOGRAFÍA
- Louis, David N., et al. cIMPACT-NOW update 1: not otherwise specified (NOS) and not elsewhere classified (NEC). Acta neuropathologica, 2018, vol. 135, no 3, p. 481-484.
- Sturm, Dominik; Pfister, Stefan M.; Jones, David TW. Pediatric gliomas: current concepts on diagnosis, biology, and clinical management. Journal of Clinical Oncology, 2017, vol. 35, no 21, p. 2370-2377.