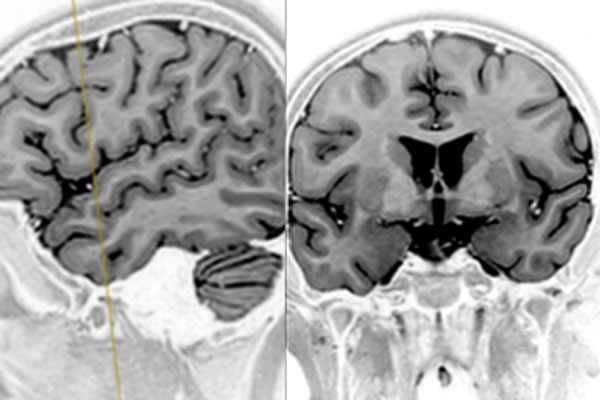
Figura 1. RM. Se observa área gliótica inespecífica en la sustancia blanca subcortical frontal derecha, sin identificarse una lesión estructural definida.
Displasia cortical leve con proliferación oligodendroglial (MOGHE) como presentación de epilepsia farmacorresistente en la infancia: reporte de un caso
Lisandro Ocampo,1 Mario Arturo Alonso Vanegas,2 Eduardo de Jesús Quintero López2
1. Servicio de Neurocirugía, Hospital Churruca-Visca, Ciudad de Buenos Aires, Argentina
2. Centro Internacional de Cirugía de Epilepsia, Servicio de Neurocirugía, HMG Coyoacán-San Ángel Inn, Ciudad de México, México
Recibido: 20/08/2025 Aceptado: 15/11/2025
EMAILS
Mario Arturo Alonso Vanegas: alonsomario@hotmail.com
Eduardo de Jesús Quintero López: drequintero@hotmail.com
Los autores no declaran conflicto de interés
Los autores no declaran financiamiento.
Este es un artículo de acceso abierto bajo la licencia CC BY-NC https://creativecommons.org/licenses/by-nc/4.0/
Palabras clave: Epilepsia del lóbulo frontal. Epilepsia farmacorresistente. Malformación del desarrollo cortical. MOGHE
Mild cortical dysplasia with oligodendroglial hyperplasia (MOGHE) presenting as drug-resistant epilepsy in childhood: a case report
ABSTRACT
Background: epilepsy affects approximately 65 million people worldwide, of whom 20–30% develop drug-resistant epilepsy, defined by the International League Against Epilepsy as the failure to achieve seizure control after the adequate use of at least two appropriately selected and tolerated antiseizure medications. This condition has a significant impact on quality of life and represents a major public health challenge, particularly in the pediatric population, where the identification of rare etiologies is essential.
Objectives: to report an infrequent case of drug-resistant epilepsy in the pediatric population and to optimize diagnostic and therapeutic processes for these conditions.
Case description: 7-year-old female patient with seizure onset at 5 years of age, initially presenting with generalized tonic-clonic seizures and subsequently evolving to focal seizures with secondary generalization. The patient developed drug-resistant epilepsy, experiencing up to three seizures per week despite multiple therapeutic regimens. Video-electroencephalography demonstrated predominant epileptiform activity in the right fronto-centro-temporal regions, while magnetic resonance imaging revealed nonspecific findings consistent with right frontal subcortical gliosis.
Surgery: given the clinical and electrophysiological concordance, surgical treatment was indicated. A right frontal craniotomy with targeted resection of the fronto-opercular region was performed. Histopathological analysis confirmed the diagnosis of mild malformation of cortical development with oligodendroglial hyperplasia associated with epilepsy (MOGHE). The patient had an uncomplicated postoperative course and remained seizure-free without neurological deficits during follow-up.
Conclusion: MOGHE represents a rare and diagnostically challenging etiology of pediatric drug-resistant epilepsy due to its limited expression on neuroimaging studies. The reporting of new cases contributes to improved recognition of this entity and supports the role of surgery as an effective therapeutic option in selected patients.
Keywords: Drug-resistant epilepsy. Frontal lobe epilepsy. Malformation of cortical development. Oligodendroglial hyperplasia associated with epilepsy (MOGHE)
INTRODUCCION
En la actualidad, se estima que aproximadamente 65000000 de personas padecen epilepsia a nivel mundial, lo que la convierte en una de las enfermedades neurológicas más prevalentes.(1) Se trata de una patología con un impacto significativo, asociada a discapacidad, deterioro de la calidad de vida y una elevada carga social y sanitaria.
Se calcula que entre el 20 y el 30 % de los pacientes con epilepsia presentan el tipo farmacorresistente, lo que incrementa sustancialmente la morbilidad y complejidad del manejo clínico.(2) De acuerdo con la Liga Internacional contra la Epilepsia (ILAE, las siglas por su nombre en inglés), la epilepsia farmacorresistente se define como la falta de control de las crisis tras el uso adecuado y tolerado de al menos 2 fármacos anticrisis, administrados en monoterapia o en combinación.(3,4)
La epilepsia farmacorresistente constituye un problema relevante de salud pública, dado que exige la consideración de estrategias terapéuticas alternativas, incluida la evaluación quirúrgica en casos seleccionados. En este contexto, el reconocimiento de etiologías poco frecuentes y de reciente descripción resulta fundamental, ya que muchas de ellas pueden pasar inadvertidas en la práctica clínica habitual debido a su escasa expresión en los estudios de neuroimagen y el limitado conocimiento disponible en la literatura médica.(5,6)
OBJETIVOS
Reportar un caso infrecuente de epilepsia farmacorresistente en la población pediátrica y optimizar los procesos de diagnóstico y tratamiento de dichas patologías.
DESCRIPCIÓN DEL CASO
Paciente de sexo femenino, de 7 años, sin antecedentes perinatales de relevancia. Nació por cesárea sin complicaciones y presentó un neurodesarrollo normal acorde a la edad. Comenzó con crisis epilépticas a los 5 años, manifestadas inicialmente como crisis tonicoclónicas generalizadas. Posteriormente, evolucionaron hacia crisis focales sin alteración de la conciencia, comprometiendo hemicara y miembros superiores izquierdos, con generalización secundaria a crisis tonicoclónicas bilaterales.
La paciente presentaba una frecuencia aproximada de 3 crisis semanales, con una duración máxima de 2 minutos y un período postictal de aproximadamente 20 minutos, caracterizado por afasia transitoria, sialorrea y letargo, con recuperación neurológica completa posterior. Recibió múltiples esquemas de tratamiento con fármacos anticrisis; el último consistió en lacosamida 100 mg cada 12 horas y oxcarbazepina en 3 dosis de 300–450–450 mg diarios. El período libre de crisis más prolongado fue de 120 días. Fue derivada a la consulta especializada por su neurólogo tratante debido a la recurrencia de crisis convulsivas sin respuesta al tratamiento farmacológico instaurado. Al ingreso, el examen físico y neurológico fue normal. Se realizó un videoelectroencefalograma (VEEG) durante el cual se registraron 2 crisis con compromiso de hemicara y hemicuerpo izquierdos, de características clínicas concordantes con las previamente detalladas. El estudio interictal evidenció un patrón eléctrico caracterizado por ondas agudas en oposición de fase y complejos punta-onda centrales bilaterales, con claro predominio derecho. Durante el período ictal, se observaron ondas agudas frontocentrales derechas con generalización secundaria, seguidas de actividad polipunta fronto-centro-temporal bilateral que evolucionó a complejos polipunta-onda en las mismas regiones. Se efectuó una resonancia magnética (RM) cerebral volumétrica, que mostró áreas de gliosis inespecíficas en la sustancia blanca subcortical frontal derecha, sin identificarse una lesión estructural definida (Figura 1). Además, la RM-PET cerebral exhibió una disminución del metabolismo a nivel del hipocampo y del giro parahipocampal izquierdos, sin otras áreas de hipometabolismo significativas.(5)
Figura 1. RM. Se observa área gliótica inespecífica en la sustancia blanca subcortical frontal derecha, sin identificarse una lesión estructural definida.
INTERVENCIÓN
El caso fue evaluado de manera interdisciplinaria en conjunto con los servicios de Neurología y Neurofisiología. Se consideró que los hallazgos semiológicos y electrofisiológicos presentaban una elevada concordancia en la localización del foco epileptógeno, sugiriendo su origen en la corteza frontal del hemisferio derecho. Si bien los estudios de resonancia magnética no evidenciaron una lesión estructural claramente definida, permitieron delimitar un área sospechosa en la región frontoopercular derecha. En este contexto, y dada la epilepsia farmacorresistente, se decidió proceder con la resección quirúrgica dirigida.(6)
La intervención quirúrgica se efectuó mediante un abordaje hemicoronal derecho, con craneotomía frontal derecha centrada en el área previamente identificada. Tras la exposición cortical, se reconoció la región frontoopercular, y se procedió a la resección del tejido sospechoso. Se obtuvo una pieza quirúrgica de aproximadamente 3 x 2 x 2 cm, la cual fue enviada para estudio anatomopatológico diferido.
El análisis histopatológico evidenció la presencia de abundantes neuronas heterotópicas en la sustancia blanca profunda, identificadas tanto por criterios morfológicos como mediante inmunohistoquímica positiva para NeuN y MAP2. Asimismo, se observó un aumento en la densidad celular de oligodendrocitos inmunorreactivos para Olig2, asociado a gliosis y a la presencia de astrocitos reactivos en la corteza suprayacente (Figura 2). En conjunto, los hallazgos morfológicos e inmunohistoquímicos fueron compatibles con una malformación leve del desarrollo con displasia cortical leve con proliferación oligodendroglial asociada a epilepsia (“Mild malformation of cortical development with Oligodendrogial hyperplasia and Epilepsy”, MOGHE).(6)
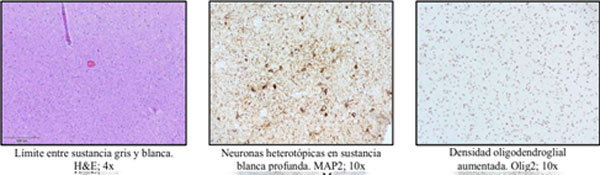
Figura 2. Informe anatomopatológico con características histopatológicas de MOGHE.
La paciente cursó el postoperatorio inmediato sin complicaciones y sin presentar déficit neurológico agregado. Se observó, además, la desaparición completa de las crisis epilépticas. Al momento del alta hospitalaria, continuó con tratamiento farmacológico consistente en lacosamida 100 mg cada 12 horas y oxcarbazepina 450 mg cada 8 horas. En los controles ambulatorios posteriores, la madre refirió la aparición de efectos adversos tras la administración de la medicación, caracterizados por mareos y diplopía. Dado que dichos síntomas fueron interpretados como secundarios al tratamiento anticonvulsivante, se decidió un descenso progresivo de la dosis de oxcarbazepina, con resolución completa de la sintomatología.
La resonancia magnética cerebral de control mostró una adecuada evolución postquirúrgica, sin hallazgos patológicos relevantes (Figura 3). Actualmente, luego de un año de seguimiento clínico, la paciente se mantiene sin déficit neurológico y libre de crisis epilépticas.
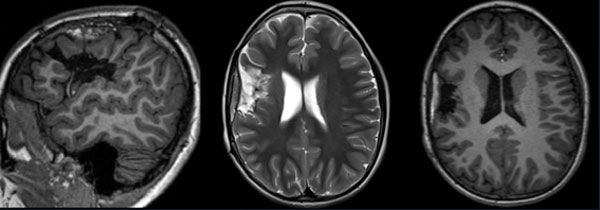
Figura 3. RM postoperatoria con resección de área de interés frontoopercular; sin complicaciones.
DISCUSIÓN
Las etiologías de la epilepsia farmacorresistente en la población pediátrica son heterogéneas e incluyen lesiones estructurales, síndromes genéticos, trastornos metabólicos, infecciones del sistema nervioso central y epilepsias de probable origen inmunomediado, entre otras. En este contexto, el diagnóstico etiológico preciso requiere una evaluación integral y sistemática de cada caso, basada en la correlación clínico-electrofisiológica, imagenológica y, en situaciones seleccionadas, anatomopatológica.(2,4)
La malformación o displasia leve del desarrollo cortical con hiperplasia oligodendroglial asociada a epilepsia (MOGHE) constituye una causa infrecuente y de descripción relativamente reciente de epilepsia focal farmacorresistente. Se trata de una entidad histopatológica que ha sido vinculada a variantes del gen SLC35A2 y cuya expresión clínica puede ser variable, incluidos espasmos epilépticos, crisis tonicoclónicas, crisis hipercinéticas y compromiso cognitivo o del neurodesarrollo.(1,5,6)
Desde el punto de vista diagnóstico, una de las principales dificultades de la entidad radica en su escasa expresión en los estudios de imágenes convencionales. Esta limitada representación favorece su confusión con displasias corticales focales u otras encefalopatías epilépticas, especialmente antes de la resección quirúrgica y del análisis histopatológico definitivo.(5) Sin embargo, una vez confirmado el diagnóstico, el pronóstico suele ser más favorable en comparación con otras etiologías estructurales de epilepsia farmacorresistente.(1)
Las series publicadas señalan el lóbulo frontal como la localización más frecuentemente comprometida, hallazgo que resultó concordante con el caso aquí presentado. Asimismo, los estudios por resonancia magnética suelen mostrar hallazgos inespecíficos, tales como borramiento de la diferenciación sustancia gris–blanca e hiperintensidades corticales o subcorticales en secuencias T2 y FLAIR, sin permitir una identificación concluyente de una malformación del desarrollo cortical.(1,5)
Diversos factores han sido asociados con un mejor pronóstico quirúrgico en pacientes con MOGHE, entre ellos el inicio temprano de las crisis, una edad menor al momento de la cirugía y la presencia de patrones epileptiformes interictales bien delimitados en el electroencefalograma, más allá de la mera identificación de la malformación en el estudio anatomopatológico.(1,6) En este sentido, también se han descripto diferentes fenotipos clínicos según la edad de presentación: en pacientes menores de 10 años se ha observado un mayor compromiso cognitivo, pero una menor duración de la epilepsia previa a la cirugía, lo que se ha asociado a mejores resultados postoperatorios en comparación con pacientes mayores o adultos.(1,6)
Por último, la fuerte asociación entre MOGHE y variantes del SLC35A2 permitió establecer que aproximadamente el 50 % de los pacientes con este diagnóstico presenten mosaicismo para dicho gen, lo que sugiere un rol patogénico central de esta alteración genética en el desarrollo de la entidad.(1,5,6)
CONCLUSIÓN
La epilepsia farmacorresistente en la infancia asociada a MOGHE continúa representando un desafío diagnóstico y terapéutico, en gran medida debido a la limitada evidencia científica disponible. Esta situación se relaciona con el carácter reciente de su descripción, el conocimiento aún incompleto de la entidad en la comunidad médica y la heterogeneidad genética y molecular observada en los pacientes afectados, factores que influyen de manera significativa en el pronóstico.
La identificación y comunicación sistemática de nuevos casos, junto con una adecuada caracterización histopatológica y genotípica, resultan fundamentales para ampliar el conocimiento sobre esta entidad y optimizar las estrategias terapéuticas, con el objetivo de ofrecer un abordaje personalizado y mejorar los resultados clínicos en esta población.
Contribuciones de autoría
Conceptualización, Metodología, Recursos y Supervisión: Mario Arturo Alonso Vanegas. Curación de datos, Análisis formal, Investigación, Software, Visualización, Redacción - borrador original y Redacción - revisión y edición: Lisandro Ocampo. Administración del proyecto y Validación: Eduardo de Jesús Quintero López. Adquisición de fondos: no aplica para este trabajo.
BIBLIOGRAFÍA
1. Shreya S, Manan S, Bhavin P. Epilepsy and its driving forces: understanding the significance behind epileptical pathogenesis. Gujarat Technological University (GTU), Ahmadabad, India. 2025. Disponible en: https://arxiv.org/html/2502.16144v1
2. Yixin Z, Shijia C, Zhenghan J. Mild malformation of cortical development with oligodendroglial hyperplasia and epilepsy: a systematic review. Neurlog Genet. 2025 Jan 31;11(1):e200240. Doi:10.1212/NXG.0000000000200240
3. Menon RN, Cross JH. Childhood epilepsy. Lancet. 2025 Aug 9;406(10503):636-49. Doi: 10.1016/S0140-6736(25)00773-1
4. Kwan P, Arzimanoglou A, Berg AT, Brodie MJ, Hauser WA, Mathern G, y col. Definition of drug-resistant epilepsy: Consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia. 2010 Jun;51(6):1069-77. Doi: 10.1111/j.1528-1167.2009.02397.x
5. Arizono E, Hori T, Saito T, Shimozawa N, Okanishi T, Baba S, y col. MRI detection of mild malformation of cortical development with oligodendroglial hyperplasia and epilepsy (MOGHE). Epilepsy Behav Rep. 2024;24:101826. Doi: 10.1016/j.ebr.2024.101826
6. Lamberink HJ, Coras R, Blümcke I, Hamer HM, Bast T, Elger CE, y col. Frontal disconnection for treating mild malformation of cortical development with oligodendroglial hyperplasia and epilepsy (MOGHE). J Vis Exp. 2024;(209):e66970. Doi: 10.3791/66970
El caso presentado aporta claridad sobre una entidad aún poco reconocida en la práctica cotidiana como es el MOGHE, cuya principal dificultad radica en su escasa expresión en imágenes y su confirmación esencialmente histopatológica.
La verdadera fortaleza de este trabajo reside en la correlación clínico-electrofisiológica que sostuvo la indicación quirúrgica, incluso frente a hallazgos radiológicos inespecíficos. Este punto es crucial: nos recuerda que, en la epilepsia farmacorresistente pediátrica, la decisión no debe ser resonancia-dependiente. Cuando la semiología y el video-EEG hablan con claridad, la cirugía se consolida como una estrategia eficaz para etiologías sutiles, pero potencialmente curables. Comunicar estos casos es fundamental para afinar nuestra sospecha diagnóstica y no dejar a estos pacientes fuera de una oportunidad terapéutica definitiva.